uORF-Tools¶
Introduction¶
uORF-Tools are a workflow and a collection of tools for the analysis of Upstream Open Reading Frames (short uORFs). The workflow is based on the workflow management system snakemake and handles installation of all dependencies via bioconda [GruningDSjodin+17], as well as all processings steps. The source code of uORF-Tools is open source and available under the License. Installation and basic usage is described below.
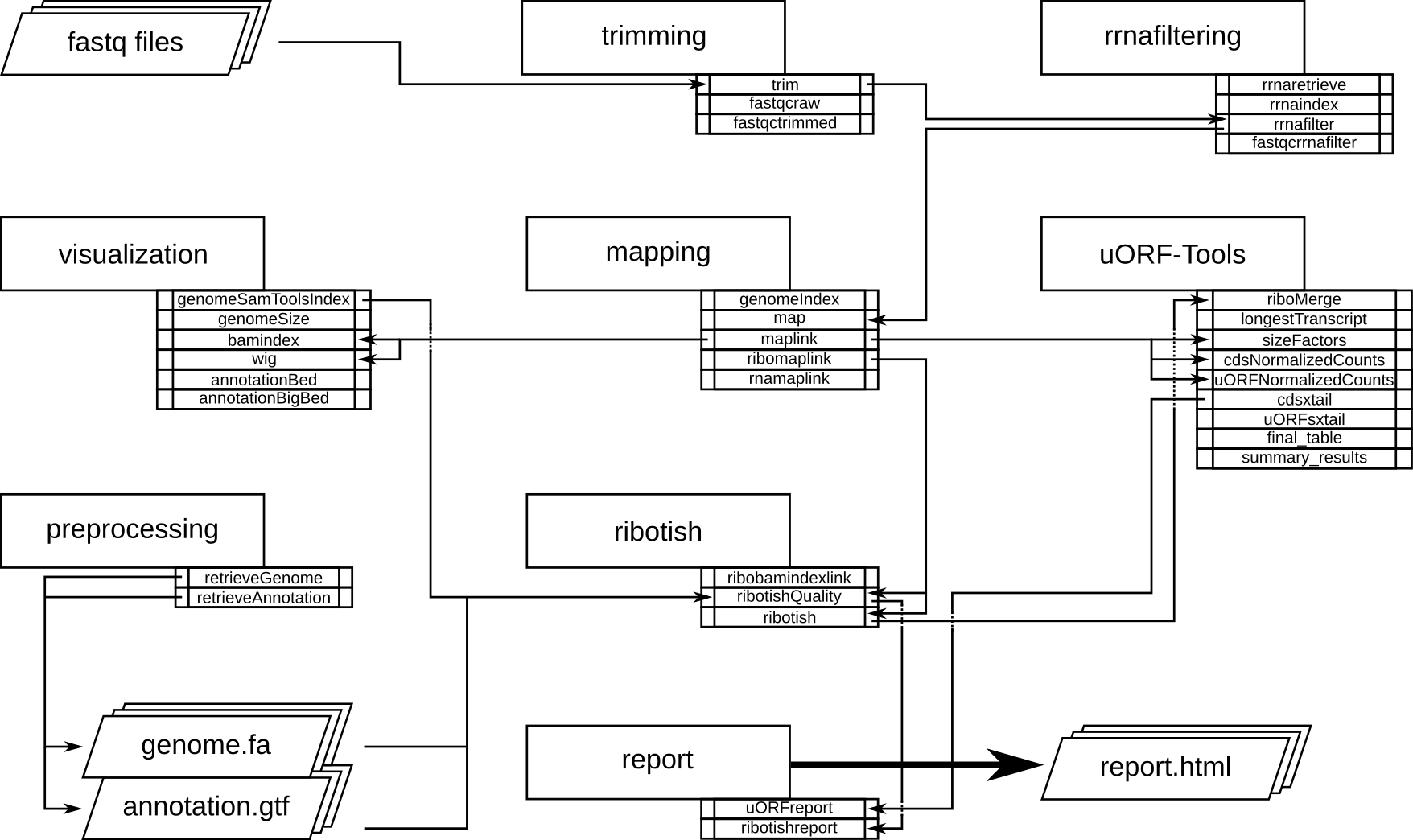
Program flowchart¶
The following flowchart describes the processing steps of the workflow and how they are connected. there is a variant of the workflow accepting a preprocessed uORF-annotation file, to skip the time consuming ribotish step for reruns of the workflow.
Directory table¶
The output is written to a directory structure that corresponds to the workflow steps, you can decide at the bedginning of the workflow whether you want to keep the intermediary files (default) or only the final result.
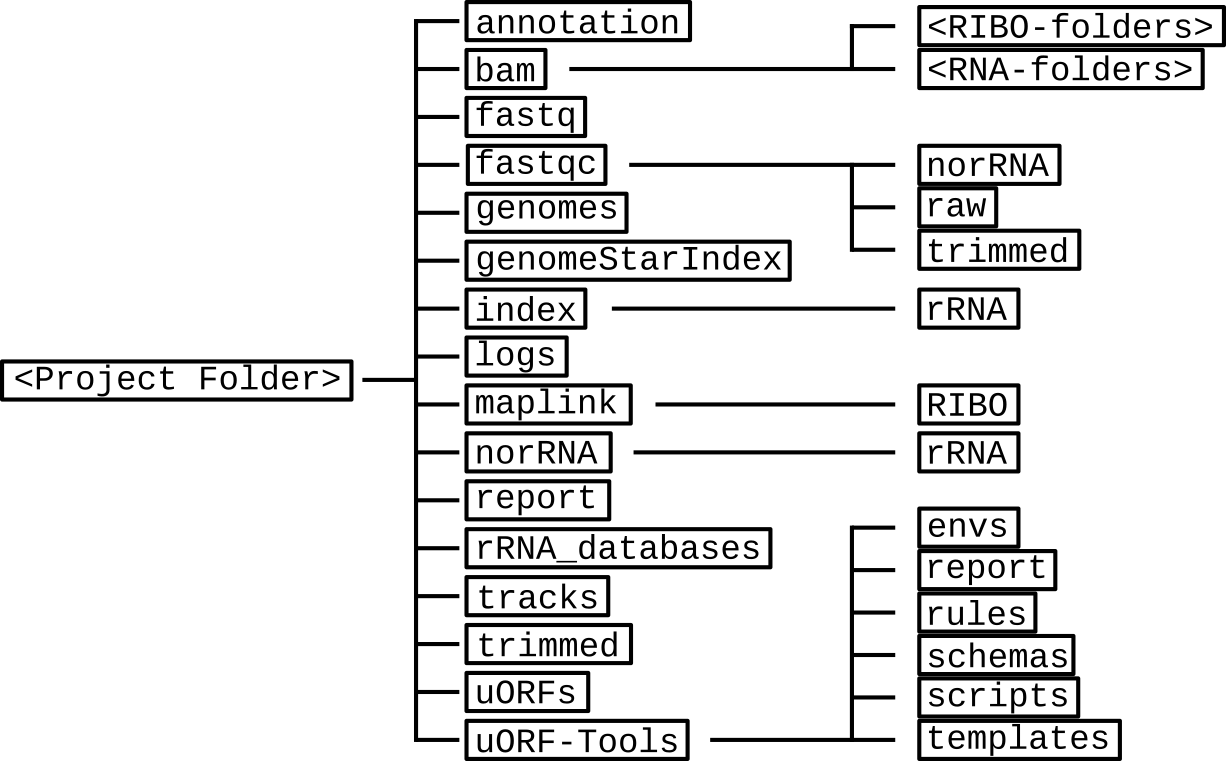
- annotation: contains the processed user-provided annotation file with genomic features.Contents: annotation.gtf
- bam: contains a subfolder for each input .fastq file. These subfolders contain the .bam files created using STAR.Contents: Aligned.sortedByCoord.out.bam, Log.final.out, Log.out, Log.progress.out, SJ.out.tab
- fastq: contains the user-provided .fastq files used as input.Contents: <method-condition-replicate>.fastq
- fastqc: contains the result files of a quality control for the input .fastq files, using different processing methods:
- norRNA: quality control after removal of the rRNA.
- raw: quality control for the unprocessed data.
- trimmed: quality control after removal of the adapter sequences.
Contents: <method-condition-replicate>_fastqc.zip, <method-condition-replicate>-<subfolderName>.html - genomes: contains the genome file, as well as an according index and sizes file.Contents: genome.fa, genome.fa.fai, sizes.genome
- genomeStarIndex: contains the files created by STAR during the creation of a genome index.Contents: chrLength.txt, chrNameLength.txt, chrStart.txt, chrName.txt, exonGeTrInfo.tab, exonInfo.tab, genInfo.tab, Genome, genomeParameters.txt, SA, SAindex, sjdbList.fromGTF.out.tab, transcriptInfo.tab, sjdbInfo.txt, sjdbList.out.tab
- index: contains an index for the rRNA databases created using indexdb_rna.Contents: <database-ID>.bursttrie_0.dat, <database-ID$>.kmer_0.dat, <database-ID>.pos_0.dat, <database-ID>.stats
- logs: contains log files for each step of the workflow.Contents: <rule>.o<jobID>, <methods>.log
- maplink: contains soft links to the .bam files and an according index.
- RIBO: contains soft links to the .bam and .bam.bai files for RIBO and corresponding parameter files (.para.py).
Contents: <method-condition-replicate>.bam.bai, RIBO/<condition-replicate>.bam.para.py - norRNA: contains processed .fastq files, where the rRNA has been removed.
- rRNA: contains the reject.fastq which specifies the rRNA reads that are removed.
Contents: <method-condition-replicate>.fastq, rRNA/reject.fastq - report: contains .jpg plots for the report.html.Contents: <condtion-replicate>-qual.jpg, xtail_cds_fc.jpg, xtail_cds_r.jpg, xtail_uORFs_fc.jpg, xtail_uORFs_r.jpg
- rRNA_databases: contains the known annotated rRNA sequences for filtering.Contents: <database-ID>.fasta
- tracks: contains BED (.bed), wig (.wig) and bigWig (.bw) files for visualizing tracks in a genome browser.Contents: annotation.bb, annotation.bed, annotation.bed6, annotationNScore.bed6, annotation-woGenes.gtf, <method-condition-replicate>.bw, <method-condition-replicate>.wig
- trimmed: contains processed .fastq files, where the adapter sequences have been trimmed.Contents: <method-condition-replicate>.fastq
- uORFs: contains the main output of the workflow.
- uORFs_regulation.tsv: table summarizing the predicted uORFs with their regulation on the main ORF.
- merged_uORFs.bed: genome browser track with predicted uORFs.
- processing_summary.tsv: table indicating the lost reads per processing step.
Contents: longest_protein_coding_transcripts.gtf, merged_uORFs.bed, merged_uORFs.csv, norm_CDS_reads.csv, norm_uORFs_reads.csv, sfactors_lprot.csv, processing_summary.tsv, uORF_regulation.tsv, xtail_cds.csv, xtail_cds_fc.pdf, xtail_cds_r.pdf, xtail_uORFs.csv, xtail_uORFs_fc.pdf, xtail_uORFs_r.pdf uORF-Tools: contains the workflow tools.
- envs: conda environment files (.yaml).
- report: restructuredText files for the report (.rst).
- rules: the snakemake rules.
- schemas: validation templates for input files
- scripts: scripts used by the snakemake workflow.
- templates: templates for the config.yaml and the samples.tsv.
Installation¶
We recommend to install uORF-Tools with all dependencies via conda. Once you have conda installed simply type:
conda create -c bioconda -c conda-forge -n uORF-Tools snakemake
source activate uORF-Tools
Usage¶
Using the workflow requires the uORF-Tools, a genome sequence (.fasta), an annotation file (.gtf) and the sequencing results files (.fastq). We recommend retrieving both the genome and the annotation files for mouse and human from GENCODE [HFG+12] and for other species from Ensembl Genomes [ZAA+18]. The usage of the workflow is first described in general, while a detailed example applied to an example dataset is described here: example-workflow.
Retrieve uORF-Tools¶
The first step is downloading the latest version of uORF-Tools from Github. Open your terminal and create a new directory for your workflow and change into it.
mkdir uORFflow; cd uORFflow;
Note
All following commands assume that you are located in the workflow folder
Now download and unpack the latest version of the uORF-Tools by entering the following commands:
wget https://github.com/anibunny12/uORF-Tools/archive/1.0.1.tar.gz
tar -xzf 1.0.1.tar.gz; mv uORF-Tools-1.0.1 uORF-Tools; rm 1.0.1.tar.gz;
The uORF-Tools are now located in a subdirectory of your workflow.
Prepare input files¶
If the genome and the annotation file are compressed, extract them using gunzip or any other decompression tool.
gunzip <genomeFile>.fa.gz
gunzip <annotationFile>.gtf.gz
Copy or move the genome and the annotation file into the workflow folder and name them genome.fa and annotation.gtf.
mv <genomeFile>.fa genome.fa
mv <annotationFile>.gtf annotation.gtf
Create a folder fastq/ and move or copy all of your compressed fastq files into the folder. .. note:: Ensure that you compress the fastq files. The workflow expects compressed files and it saves a lot of disk space.
mkdir fastq
mv *.fastq.gz fastq/
Now copy the templates of the sample sheet and the configuration file into the uORF-Tools folder.
cp uORF-Tools/templates/samples.tsv uORF-Tools/
cp uORF-Tools/templates/config.yaml uORF-Tools/
Next, customize the config.yaml. It contains the following variables:
- taxonomy Specify the taxonomic group of the used organism in order to ensure the correct removal of reads mapping to ribosomal genes (Eukarya, Bacteria, Archea).
- adapter Specify the adapter sequence to be used. If not set, Trim galore will try to determine it automatically.
- samples The location of the samples sheet created in the previous step.
- genomeindexpath If the STAR genome index was already precomputed, you can specify the path to the files here, in order to avoid recomputation.
- uorfannotationpath If the uORF-file was already precomputed, you can specify the path to the files here, in order to avoid recomputation.
Now edit the sample sheet corresponding to your project. It contains the following variables:
- method Indicates the method used for this project. RIBO for ribosome profiling or RNA for RNA-seq.
- condition Indicates the applied condition (A, B / CTRL, TREAT). Please ensure that you put the control before the treatment alphabetically (e.g. A: Control B: Treatment or CTRL: Control, TREAT: Treatment)
- replicate ID used to distinguish between the different replicates (e.g. 1,2, …)
- fastqFile Indicates the according fastq file for a given sample.
As seen in the samples.tsv template:
| method | condition | replicate | fastqFile |
|---|---|---|---|
| RIBO | A | 1 | fastq/FP-ctrl-1-2.fastq.gz |
| RIBO | B | 1 | fastq/FP-treat-1-2.fastq.gz |
| RNA | A | 1 | fastq/Total-ctrl-1-2.fastq.gz |
| RNA | B | 1 | fastq/Total-treat-1-2.fastq.gz |
Executing the workflow¶
The workflow will first retrieve all required programs and install them. Then it will derive the necessary computation step depending on your input files. You will receive continuous updates about the progress of the workflow execution. Log files of the individual steps will be written to the logs subdirectory and are named according to the workflow step. The intermediary output of the different workflow steps are written to directories as shown in the directory table.
Run the workflow locally¶
Use the following steps when you plan to execute the workflow on a single server or workstation. Please be aware that some steps of the workflow require a lot of memory, specifically for eukaryotic species.
snakemake --use-conda -s uORF-Tools/Snakefile --configfile uORF-Tools/config.yaml --directory ${PWD} -j 20 --latency-wait 60
Run Snakemake in a cluster environment¶
Use the following steps if you are executing the workflow via a queuing system. Edit the configuration file cluster.yaml according to your queuing system setup and cluster hardware. The following system call shows the usage with Grid Engine:
snakemake --use-conda -s uORF-Tools/Snakefile --configfile uORF-Tools/config.yaml --directory ${PWD} -j 20 --cluster-config uORF-Tools/cluster.yaml
Report¶
Using any of the presented methods, this will run the workflow on our dataset and create the desired output files. Once the workflow has finished, we can request an automatically generated report.html file using the following command:
snakemake --report report.html
References¶
| [GruningDSjodin+17] | Björn Grüning, Ryan Dale, Andreas Sjödin, Jillian Rowe, Brad A. Chapman, Christopher H. Tomkins-Tinch, Renan Valieris, and Johannes Köster. Bioconda: a sustainable and comprehensive software distribution for the life sciences. bioRxiv, 2017. URL: https://www.biorxiv.org/content/early/2017/10/27/207092, arXiv:https://www.biorxiv.org/content/early/2017/10/27/207092.full.pdf, doi:10.1101/207092. |
| [HFG+12] | J. Harrow, A. Frankish, J. M. Gonzalez, E. Tapanari, M. Diekhans, F. Kokocinski, B. L. Aken, D. Barrell, A. Zadissa, S. Searle, I. Barnes, A. Bignell, V. Boychenko, T. Hunt, M. Kay, G. Mukherjee, J. Rajan, G. Despacio-Reyes, G. Saunders, C. Steward, R. Harte, M. Lin, C. Howald, A. Tanzer, T. Derrien, J. Chrast, N. Walters, S. Balasubramanian, B. Pei, M. Tress, J. M. Rodriguez, I. Ezkurdia, J. van Baren, M. Brent, D. Haussler, M. Kellis, A. Valencia, A. Reymond, M. Gerstein, R. Guigo, and T. J. Hubbard. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res., 22(9):1760–1774, Sep 2012. |
| [LSS11] | R. Leinonen, H. Sugawara, and M. Shumway. The sequence read archive. Nucleic Acids Res., 39(Database issue):19–21, Jan 2011. |
| [SAA+17] | Nicole Silvester, Blaise Alako, Clara Amid, Ana Cerdeño-Tarrága, Laura Clarke, Iain Cleland, Peter W Harrison, Suran Jayathilaka, Simon Kay, Thomas Keane, and others. The european nucleotide archive in 2017. Nucleic acids research, 46(D1):D36–D40, 2017. |
| [ZAA+18] | Daniel R Zerbino, Premanand Achuthan, Wasiu Akanni, M Ridwan Amode, Daniel Barrell, Jyothish Bhai, Konstantinos Billis, Carla Cummins, Astrid Gall, Carlos García Girón, Laurent Gil, Leo Gordon, Leanne Haggerty, Erin Haskell, Thibaut Hourlier, Osagie G Izuogu, Sophie H Janacek, Thomas Juettemann, Jimmy Kiang To, Matthew R Laird, Ilias Lavidas, Zhicheng Liu, Jane E Loveland, Thomas Maurel, William McLaren, Benjamin Moore, Jonathan Mudge, Daniel N Murphy, Victoria Newman, Michael Nuhn, Denye Ogeh, Chuang Kee Ong, Anne Parker, Mateus Patricio, Harpreet Singh Riat, Helen Schuilenburg, Dan Sheppard, Helen Sparrow, Kieron Taylor, Anja Thormann, Alessandro Vullo, Brandon Walts, Amonida Zadissa, Adam Frankish, Sarah E Hunt, Myrto Kostadima, Nicholas Langridge, Fergal J Martin, Matthieu Muffato, Emily Perry, Magali Ruffier, Dan M Staines, Stephen J Trevanion, Bronwen L Aken, Fiona Cunningham, Andrew Yates, and Paul Flicek. Ensembl 2018. Nucleic Acids Research, 46(D1):D754–D761, 2018. URL: http://dx.doi.org/10.1093/nar/gkx1098, arXiv:/oup/backfile/content_public/journal/nar/46/d1/10.1093_nar_gkx1098/2/gkx1098.pdf, doi:10.1093/nar/gkx1098. |