uORF-Tools¶
Introduction¶
uORF-Tools is a workflow and a collection of tools for the analysis of Upstream Open Reading Frames (short uORFs). The workflow is based on the workflow management system snakemake and handles installation of all dependencies via bioconda [GruningDSjodin+17], as well as all processings steps. The source code of uORF-Tools is open source and available under the License GNU General Public License 3. Installation and basic usage is described below.
Note
For a detailed step by step tutorial of our workflow on a sample dataset, please refer to our example-workflow.
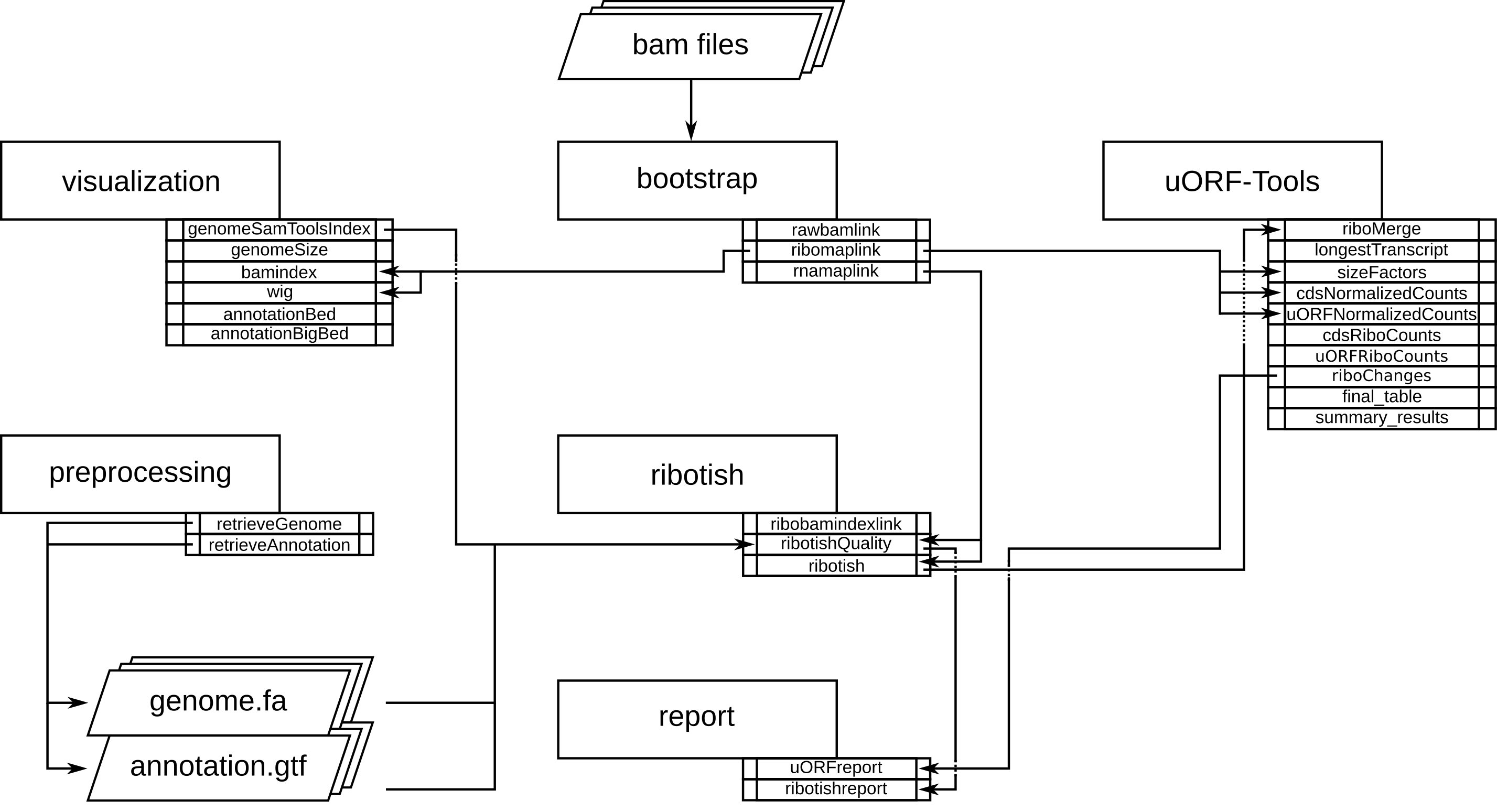
Program flowchart¶
The following flowchart describes the processing steps of the workflow and how they are connected:
Directory table¶
The output is written to a directory structure that corresponds to the workflow steps, you can decide at the beginning of the workflow whether you want to keep the intermediary files (default) or only the final result.
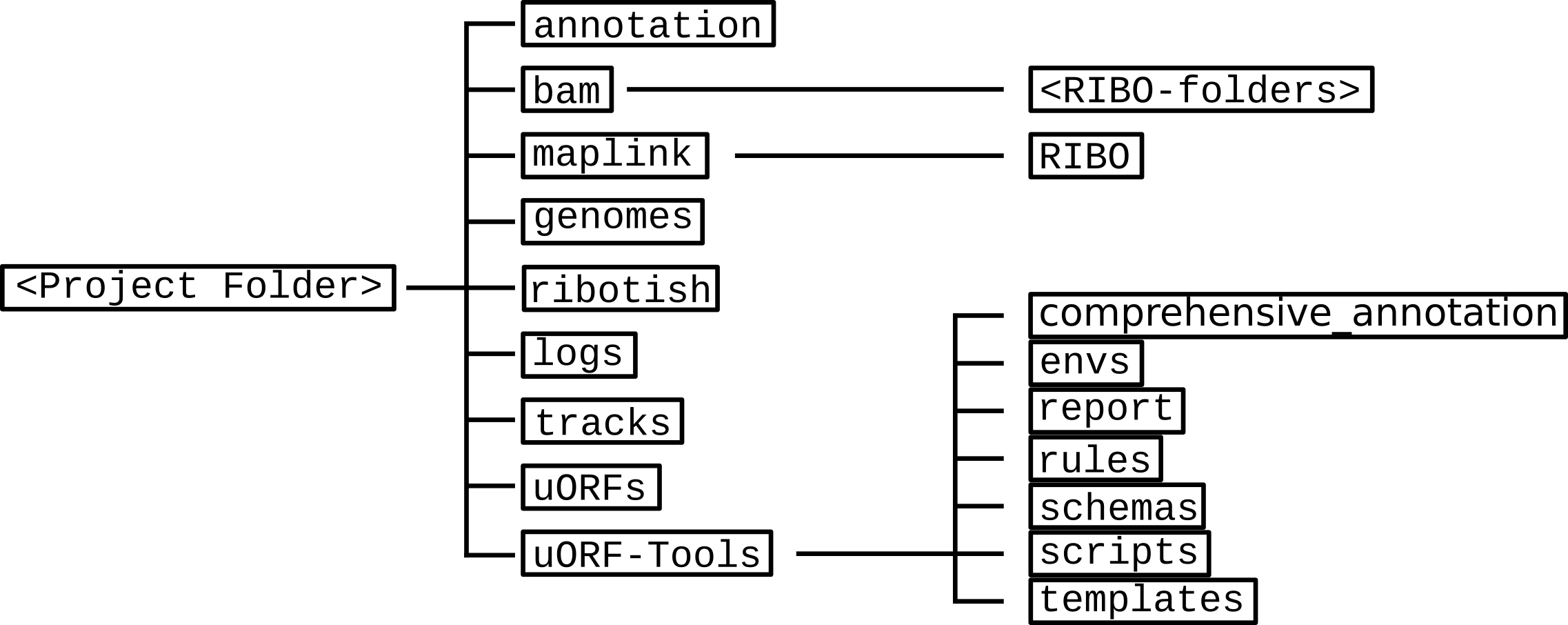
- annotation: contains the user-provided annotation file with genomic features.Contents: annotation.gtf
- bam: contains the input .bam files.Contents: <method>-<conditon>-<replicate>.bam
- genomes: contains the genome file, as well as an according index and sizes file.Contents: genome.fa, genome.fa.fai, sizes.genome
- logs: contains log files for each step of the workflow.Contents: <rule>.o<jobID>, <methods>.log
- maplink: contains soft links to the .bam files and an according index.
- RIBO: contains soft links to the .bam and .bam.bai files for RIBO and corresponding parameter files (.para.py).
Contents: <method-condition-replicate>.bam.bai, RIBO/<condition-replicate>.bam.para.py - ribotish: contains the result files of ribotish.Contents: <condition-replicate>-newORFs.tsv, <condition-replicate>-newORFs.tsv_all.txt, <condition-replicate>-qual.txt, <condition-replicate>-qual.pdf
- tracks: contains BED (.bed), wig (.wig) and bigWig (.bw) files for visualizing tracks in a genome browser.Contents: annotation.bb, annotation.bed, annotation.bed6, annotation-woGenes.gtf, <method-condition-replicate>.bw, <method-condition-replicate>.wig
- uORFs: contains the main output of the workflow.
- uORFs_regulation.tsv: table summarizing the predicted uORFs with their regulation on the main ORF.
- merged_uORFs.bed: genome browser track with predicted uORFs.
Contents: longest_protein_coding_transcripts.gtf, merged_uORFs.bed, merged_uORFs.csv, ribo_norm_CDS_reads.csv, ribo_norm_uORFs_reads.csv, ribo_raw_CDS_reads.csv, ribo_raw_uORFs_reads.csv, sfactors_lprot.csv, uORFs_regulation.tsv uORF-Tools: contains the workflow tools.
- comprehensive_annotation: an example annotation.
- envs: conda environment files (.yaml).
- report: restructuredText files for the report (.rst).
- rules: the snakemake rules.
- schemas: validation templates for input files
- scripts: scripts used by the snakemake workflow.
- templates: templates for the config.yaml and the samples.tsv.
Input/Output Files¶
This table contains explanations for each of the input/output files.
| File name | Description |
|---|---|
| annotation.gtf | user-provided annotation file with genomic features |
| genome.fa | user-provided genome file containing the genome sequence |
| genome.fa.fai | index file of the genome file |
| sizes.genome | file containing the sizes of each genome sequence in the genome file |
| <method>-<conditon>-<replicate>.bam | user-provided alignment files (or created using the preprocessing workflow) |
| <method-condition-replicate>.bam.bai | index file of the alignment files |
| <condition-replicate>.bam.para.py | parameter file generated by RiboTISH |
| <methods>.log | files containing the process log for each method |
| <condition-replicate>-newORFs.tsv | RiboTISH output file containing newly discovered ORFs (significant only) |
| <condition-replicate>-newORFs.tsv_all.txt | RiboTISH output file containing newly discovered ORFs (all) |
| <condition-replicate>-qual.txt | RiboTISH quality control report text file |
| <condition-replicate>-qual.pdf | RiboTISH quality control report file with illustrations |
| annotation.bb | input annotation in bigbed format for genome browser visualization |
| annotation.bed | input annotation in bed format for genome browser visualization |
| annotation.bed6 | input annotation in bed6 format for genome browser visualization |
| annotation-woGenes.gtf | input annotation filtered exclusively for gene features |
| <method-condition-replicate>.bw | BigWig files for visualizing data in a genome Browser |
| <method-condition-replicate>.wig | wig files for visualizing data in a genome Browser |
| uORFs_regulation.tsv | final output table including all uORFs and their mORF |
| merged_uORFs.bed | bed file containing potential ORFs for genome browser visualization |
| merged_uORFs.csv | list of potential ORFs with coordinates and mORF annotation |
| longest_protein_coding_transcripts.gtf | input annotation filtered for longest splice variant for each locus |
| ribo_raw_CDS_reads.csv | read counts for annotated ORFs |
| ribo_raw_uORFs_reads.csv | read counts for potential ORFs |
| ribo_norm_CDS_reads.csv | deseq2 normalized read counts for annotated ORFs |
| ribo_norm_uORFs_reads.csv | deseq2 normalized read counts for potential ORFs |
| sfactors_lprot.csv | deseq2 size factors for protein coding transcripts |
uORFs_regulation.tsv¶
Description for the columns present in the final output file:
- transcript_id: transcript id of the main open reading frame (mORF)
- uORF_id: id of the potential Upstream open reading frame (uORF), derived from mORF id
- Ratio: list of columns, one for each sample, with the ratio of read counts for the mORF and the uORF
- Standard deviation of changes of the ratio of the relative uORF activities of treatment vs control
- binary logarithm fold change of the ratio of the relative uORF activities of treatment vs control
Tool Parameters¶
Special characters and versions used for the most important tools. Standard input/output parameters were omitted.
| Tool | Version | Special parameters used |
|---|---|---|
| riboTISH | 0.2.1 | –longest (-v -p -b -g -f) |
| trim-galore | 0.5.0 | –phred33 -q 20 –length 15 –trim-n –suppress_warn –clip_R1 1 –dont_gzip |
| star | 2.6.1b | –genomeDir genomeStarIndex –outSAMtype BAM SortedByCoordinate –outSAMattributes All –outFilterMultimapNmax 1 –alignEndsType Extend5pOfRead1 |
| sortmerna | 2.1b | -m 4096 -a –ref <dbstring> –reads –num_alignments 1 –fastx –aligned –other |
| fastqc | 0.11.8 | |
| imagemagick | 7.0.8_15 | -density 150 -trim -quality 100 -flatten -sharpen 0x1.0 |
Requirements¶
In the following, we describe all the required files and tools needed to run our workflow.
Tools¶
miniconda3¶
As this workflow is based on the workflow management system snakemake [KosterR18], Snakemake will download all necessary dependencies via conda.
We strongly recommend installing miniconda3 with python3.7.
After downloading the miniconda3 version suiting your linux system, execute the downloaded bash file and follow the instructions given.
snakemake¶
Note
The uORF-Tools require snakemake (version==5.4.5)
The newest version of snakemake can be downloaded via conda using the following command:
$ conda create -c conda-forge -c bioconda -n snakemake snakemake==5.4.5
This creates a new conda environment called snakemake and installs snakemake into the environment. The environment can be activated using:
$ conda activate snakemake
and deactivated using:
$ conda deactivate
uORF-Tools¶
Using the workflow requires the uORF-Tools. The latest version is available on our GitHub page.
In order to run the workflow, we suggest that you download the uORF-Tools into your project directory. The following command creates an example directory and changes into it:
$ mkdir project
$ cd project
Now, download and unpack the latest version of the uORF-Tools by entering the following commands:
$ wget https://github.com/Biochemistry1-FFM/uORF-Tools/archive/3.2.1.tar.gz
$ tar -xzf 3.2.1.tar.gz; mv uORF-Tools-3.2.1 uORF-Tools; rm 3.2.1.tar.gz;
The uORF-Tools are now a subdirectory of your project directory.
Input files¶
Several input files are required in order to run our workflow, a genome sequence (.fa), an annotation file (.gtf) and the bam files (.bam).
genome.fa and annotation.gtf¶
We recommend retrieving both the genome and the annotation files for mouse and human from GENCODE [HFG+12] and for other species from Ensembl Genomes [ZAA+18].
Note
For detailed information about downloading and unpacking these files, please refer to our example-workflow.
input .bam files¶
These are the input files provided by you (the user).
“For best performance, reads should be trimmed (to ~ 29 nt RPF length) and aligned to genome using end-to-end mode (no soft-clip). Intron splicing is supported. Some attributes are needed such as NM, NH and MD. For STAR, –outSAMattributes All should be set. bam file should be sorted and indexed by samtools.” (RiboTISH requirements, see https://github.com/zhpn1024/ribotish ).
Please ensure that you move all input .bam files into a folder called bam (Located in your project folder):
$ mkdir bam
$ cp *.bam bam/
Sample sheet and configuration file¶
In order to run the uORF-Tools, you have to provide a sample sheet and a configuration file. There are templates for both files available in the uORF-Tools folder.
Copy the templates of the sample sheet and the configuration file into the uORF-Tools folder:
$ cp uORF-Tools/templates/samples.tsv uORF-Tools/
$ cp uORF-Tools/templates/config.yaml uORF-Tools/
Customize the config.yaml using your preferred editor. It contains the following variables:
- taxonomy Specify the taxonomic group of the used organism in order to ensure the correct removal of reads mapping to ribosomal genes (Eukarya, Bacteria, Archea). (Option for the preprocessing workflow)
- adapter Specify the adapter sequence to be used. If not set, Trim galore will try to determine it automatically. (Option for the preprocessing workflow)
- samples The location of the samples sheet created in the previous step.
- genomeindexpath If the STAR genome index was already precomputed, you can specify the path to the files here, in order to avoid recomputation. (Option for the preprocessing workflow)
- uorfannotationpath If a uORF-annotation file was already pre-computed, you can specify the path to the file here. Please make sure, that the file has the same format as the uORF_annotation_hg38.csv file provided in the git repo (i.e. same number of columns, same column names)
- alternativestartcodons Specify a comma separated list of alternative start codons.
Edit the sample sheet corresponding to your project. It contains the following variables:
- method Indicates the method used for this project, here RIBO for ribosome profiling.
- condition Indicates the applied condition (e.g. A, B, …).
- replicate ID used to distinguish between the different replicates (e.g. 1,2, …)
- inputFile Indicates the according bam file for a given sample.
As seen in the bam-samples.tsv template:
| method | condition | replicate | inputFile |
|---|---|---|---|
| RIBO | A | 1 | bam/RIBO-A-1.bam |
| RIBO | A | 2 | bam/RIBO-A-2.bam |
| RIBO | A | 3 | bam/RIBO-A-3.bam |
| RIBO | A | 4 | bam/RIBO-A-4.bam |
| RIBO | B | 1 | bam/RIBO-B-1.bam |
| RIBO | B | 2 | bam/RIBO-B-2.bam |
| RIBO | B | 3 | bam/RIBO-B-3.bam |
| RIBO | B | 4 | bam/RIBO-B-4.bam |
Warning
Please make sure that you have at-least two replicates for each condition!
Warning
Please ensure that you put the treatment before the control alphabetically (e.g. A: Treatment B: Control)
cluster.yaml¶
In the template folder, we provide two cluster.yaml files needed by snakemake in order to run on a cluster system:
- sge-cluster.yaml - for grid based queuing systems
- torque-cluster.yaml - for torque based queuing systems
Example-workflow¶
A detailed step by step tutorial is available at: example-workflow.
Preprocessing-workflow¶
We also provide an preprocessing workflow containing a preprocessing step, starting with fastq files. A detailed step by step tutorial is available at: preprocessing-workflow.
References¶
| [GruningDSjodin+17] | Björn Grüning, Ryan Dale, Andreas Sjödin, Jillian Rowe, Brad A. Chapman, Christopher H. Tomkins-Tinch, Renan Valieris, and Johannes Köster. Bioconda: a sustainable and comprehensive software distribution for the life sciences. bioRxiv, 2017. URL: https://www.biorxiv.org/content/early/2017/10/27/207092, arXiv:https://www.biorxiv.org/content/early/2017/10/27/207092.full.pdf, doi:10.1101/207092. |
| [HFG+12] | J. Harrow, A. Frankish, J. M. Gonzalez, E. Tapanari, M. Diekhans, F. Kokocinski, B. L. Aken, D. Barrell, A. Zadissa, S. Searle, I. Barnes, A. Bignell, V. Boychenko, T. Hunt, M. Kay, G. Mukherjee, J. Rajan, G. Despacio-Reyes, G. Saunders, C. Steward, R. Harte, M. Lin, C. Howald, A. Tanzer, T. Derrien, J. Chrast, N. Walters, S. Balasubramanian, B. Pei, M. Tress, J. M. Rodriguez, I. Ezkurdia, J. van Baren, M. Brent, D. Haussler, M. Kellis, A. Valencia, A. Reymond, M. Gerstein, R. Guigo, and T. J. Hubbard. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res., 22(9):1760–1774, Sep 2012. |
| [KosterR18] | Johannes Köster and Sven Rahmann. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics, ():bty350, 2018. URL: http://dx.doi.org/10.1093/bioinformatics/bty350, arXiv:/oup/backfile/content_public/journal/bioinformatics/pap/10.1093_bioinformatics_bty350/2/bty350.pdf, doi:10.1093/bioinformatics/bty350. |
| [ZAA+18] | Daniel R Zerbino, Premanand Achuthan, Wasiu Akanni, M Ridwan Amode, Daniel Barrell, Jyothish Bhai, Konstantinos Billis, Carla Cummins, Astrid Gall, Carlos García Girón, Laurent Gil, Leo Gordon, Leanne Haggerty, Erin Haskell, Thibaut Hourlier, Osagie G Izuogu, Sophie H Janacek, Thomas Juettemann, Jimmy Kiang To, Matthew R Laird, Ilias Lavidas, Zhicheng Liu, Jane E Loveland, Thomas Maurel, William McLaren, Benjamin Moore, Jonathan Mudge, Daniel N Murphy, Victoria Newman, Michael Nuhn, Denye Ogeh, Chuang Kee Ong, Anne Parker, Mateus Patricio, Harpreet Singh Riat, Helen Schuilenburg, Dan Sheppard, Helen Sparrow, Kieron Taylor, Anja Thormann, Alessandro Vullo, Brandon Walts, Amonida Zadissa, Adam Frankish, Sarah E Hunt, Myrto Kostadima, Nicholas Langridge, Fergal J Martin, Matthieu Muffato, Emily Perry, Magali Ruffier, Dan M Staines, Stephen J Trevanion, Bronwen L Aken, Fiona Cunningham, Andrew Yates, and Paul Flicek. Ensembl 2018. Nucleic Acids Research, 46(D1):D754–D761, 2018. URL: http://dx.doi.org/10.1093/nar/gkx1098, arXiv:/oup/backfile/content_public/journal/nar/46/d1/10.1093_nar_gkx1098/2/gkx1098.pdf, doi:10.1093/nar/gkx1098. |