Example workflow¶
The retrieval of input files and running the workflow locally and on a server cluster via a queuing system is demonstrated using an example with data available from our FTP-Server. The dataset is available under the GEO accession number GSE103719.
Note
Ensure that you have miniconda3 installed and a conda environment set-up. Please refer to the overview for details on the installation.
Setup¶
First of all, we start by creating the project directory and changing to it.
$ mkdir project
$ cd project
We then download the latest version of the uORF-Tools into the newly created project folder and unpack it.
$ wget https://github.com/Biochemistry1-FFM/uORF-Tools/archive/3.2.1.tar.gz
$ tar -xzf 3.2.1.tar.gz; mv uORF-Tools-3.2.1 uORF-Tools; rm 3.2.1.tar.gz;
Retrieve and prepare input files¶
Before starting the workflow, we have to acquire and prepare several input files. These files are the annotation file, the genome file, the bam files, the configuration file and the sample sheet.
Annotation and genome files¶
First, we want to retrieve the annotation file and the genome file. In this case, we can find both on the GENCODE [HFG+12] webpage for the human genome.
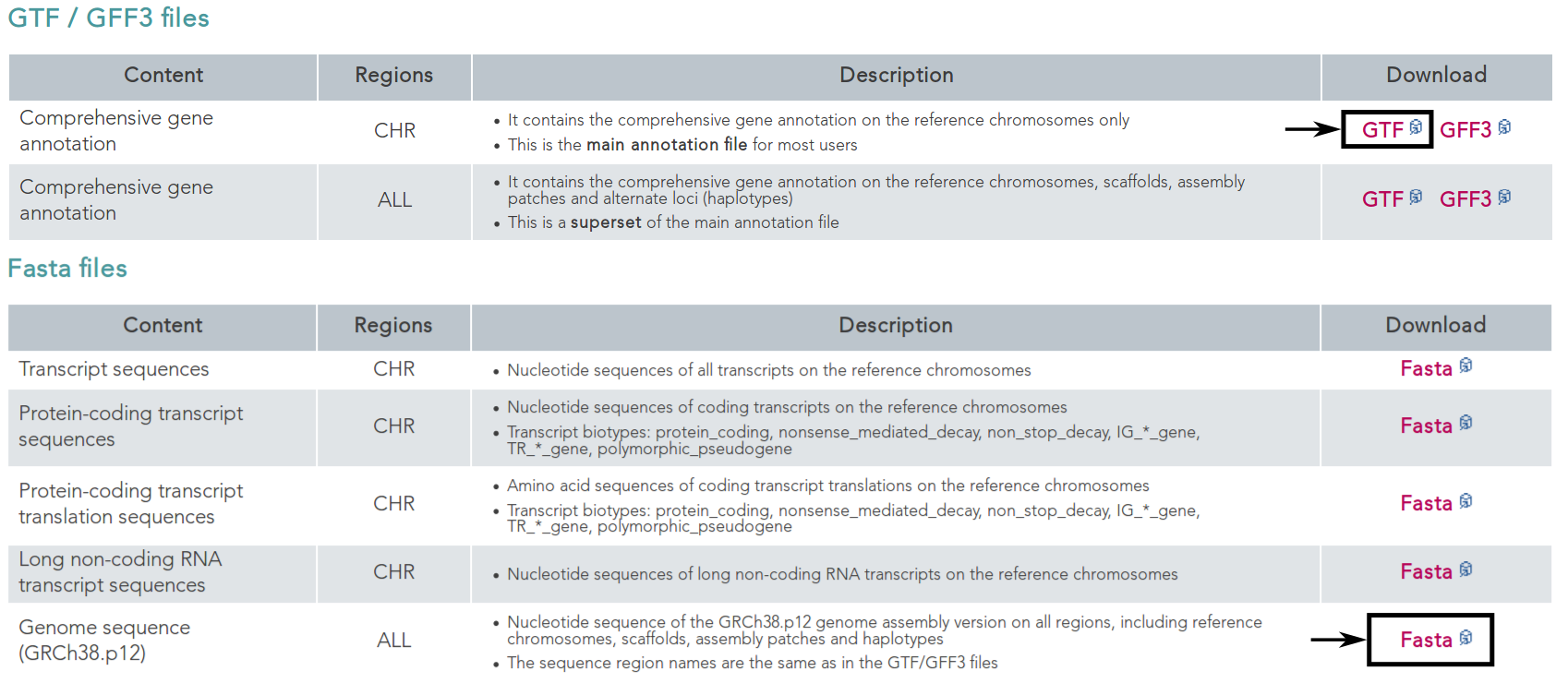
On this page, we can directly retrieve both files by clicking on the according download links next to the file descriptions. Alternatively, you can directly download them using the following commands:
$ wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_28/gencode.v28.annotation.gtf.gz && wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_28/GRCh38.p12.genome.fa.gz
Then, we are going to unpack both files.
$ gunzip gencode.v28.annotation.gtf.gz && gunzip GRCh38.p12.genome.fa.gz
Finally, we will rename these files to annotation.gtf and genome.fa.
$ mv gencode.v28.annotation.gtf annotation.gtf && mv GRCh38.p12.genome.fa genome.fa
Another webpage that provides these files is Ensembl Genomes [ZAA+18]. This usually requires searching their file system in order to find the wanted files. For this tutorial, we recommend to stick to GenCode instead.
.bam files¶
Next, we want to acquire the bam files. The bam files for the tutorial dataset can be downloaded from our FTP-Server:
Note
We provide both a .zip and a .tar.gz file. We recommend the .tar.gz file as most linux systems can decompress them via commandline by default.
$ wget ftp://biftp.informatik.uni-freiburg.de/pub/uORF-Tools/bam.tar.gz; tar -zxvf bam.tar.gz; rm bam.tar.gz;
This will create a bam folder containing all the files necessary to run the workflow. If you prefer using your own .bam files, create a bam folder and copy the files into it. Make sure that your reads were trimmed (to ~29bp length) and aligned to the genome using end-to-end alignment. The bam files need to include all SAM attributes and should be sorted using samtools.
$ mkdir bam
$ cp *.bam bam/
Sample sheet and configuration file¶
Finally, we will prepare the configuration file (config.yaml) and the sample sheet (samples.tsv). We start by copying templates for both files from the uORF-Tools/templates/ into the uORF-Tools/ folder.
$ cp uORF-Tools/templates/bam-samples.tsv uORF-Tools/
$ mv uORF-Tools/bam-samples.tsv uORF-Tools/samples.tsv
The sample file looks as follows:
| method | condition | replicate | inputFile |
|---|---|---|---|
| RIBO | A | 1 | bam/RIBO-A-1.bam |
| RIBO | A | 2 | bam/RIBO-A-2.bam |
| RIBO | A | 3 | bam/RIBO-A-3.bam |
| RIBO | A | 4 | bam/RIBO-A-4.bam |
| RIBO | B | 1 | bam/RIBO-B-1.bam |
| RIBO | B | 2 | bam/RIBO-B-2.bam |
| RIBO | B | 3 | bam/RIBO-B-3.bam |
| RIBO | B | 4 | bam/RIBO-B-4.bam |
Note
When using your own data, use any editor (vi(m), gedit, nano, atom, …) to customize the sample sheet.
Warning
Please ensure not to replace any tabulator symbols with spaces while changing this file.
Next, we are going to set up the config.yaml.
$ cp uORF-Tools/templates/config.yaml uORF-Tools/
$ vi uORF-Tools/config.yaml
This file contains the following variables:
- taxonomy Specify the taxonomic group of the used organism in order to ensure the correct removal of reads mapping to ribosomal genes (Eukarya, Bacteria, Archea). (Option for the preprocessing workflow)
- adapter Specify the adapter sequence to be used. If not set, Trim galore will try to determine it automatically. (Option for the preprocessing workflow)
- samples The location of the samples sheet created in the previous step.
- genomeindexpath If the STAR genome index was already precomputed, you can specify the path to the files here, in order to avoid recomputation. (Option for the preprocessing workflow)
- uorfannotationpath If a uORF-annotation file was already pre-computed, you can specify the path to the file here. Please make sure, that the file has the same format as the uORF_annotation_hg38.csv file provided in the git repo (i.e. same number of columns, same column names)
- alternativestartcodons Specify a comma separated list of alternative start codons.
#Taxonomy of the samples to be processed, possible are Eukarya, Bacteria, Archea
taxonomy: "Eukarya"
#Adapter sequence used
adapter: ""
samples: "uORF-Tools/samples.tsv"
genomeindexpath: ""
uorfannotationpath: ""
alternativestartcodons: ""
For this tutorial, we can keep the default values for the config.yaml. The organism analyzed in this tutorial is homo sapiens, therefore we keep the taxonomy at Eukarya. The path to samples.tsv is set correctly.
Running the workflow¶
Now that we have all the required files, we can start running the workflow, either locally or in a cluster environment.
Run the workflow locally¶
Use the following steps when you plan to execute the workflow on a single server or workstation. Please be aware that some steps of the workflow might require a lot of memory, specifically for eukaryotic species.
Navigate to the project folder containing the bam/ folder, the annotation.gtf and the genome.fa files and the uORF-Tools folder. Start the workflow locally from this folder by running:
$ snakemake --use-conda -s uORF-Tools/Snakefile --configfile uORF-Tools/config.yaml --directory ${PWD} -j 20 --latency-wait 60
Run Snakemake in a cluster environment¶
Use the following steps if you are executing the workflow via a queuing system. Edit the configuration file cluster.yaml according to your queuing system setup and cluster hardware.
Navigate to the project folder on your cluster system. Start the workflow from this folder by running (The following system call shows the usage with Grid Engine.):
$ snakemake --use-conda -s uORF-Tools/Snakefile --configfile uORF-Tools/config.yaml --directory ${PWD} -j 20 --cluster-config uORF-Tools/templates/sge-cluster.yaml
Example: Run Snakemake in a cluster environment¶
Warning
Be advised that this is a specific example, the required options may change depending on your system.
We ran the tutorial workflow in a cluster environment, specifically a TORQUE cluster environment. Therefore, we created a bash script torque.sh in our project folder.
$ vi torque.sh
Note
Please note that all arguments enclosed in <> have to be customized. This script will only work if your cluster uses the TORQUE queuing system.
We proceeded by writing the queuing script:
#!/bin/bash
#PBS -N <ProjectName>
#PBS -S /bin/bash
#PBS -q "long"
#PBS -d <PATH/ProjectFolder>
#PBS -l nodes=1:ppn=1
#PBS -o <PATH/ProjectFolder>
#PBS -j oe
cd <PATH/ProjectFolder>
source activate uORF-Tools
snakemake --latency-wait 600 --use-conda -s uORF-Tools/Snakefile --configfile uORF-Tools/config.yaml --directory ${PWD} -j 20 --cluster-config uORF-Tools/templates/torque-cluster.yaml --cluster "qsub -N {cluster.jobname} -S /bin/bash -q {cluster.qname} -d <PATH/ProjectFolder> -l {cluster.resources} -o {cluster.logoutputdir} -j oe"
We then simply submitted this job to the cluster:
$ qsub torque.sh
Using any of the presented methods, this will run the workflow on our dataset and create the desired output files.
Report¶
Once the workflow has finished, we can request an automatically generated report.html file using the following command:
$ snakemake --use-conda -s uORF-Tools/Snakefile --configfile uORF-Tools/config.yaml --report report.html
References¶
| [HFG+12] | J. Harrow, A. Frankish, J. M. Gonzalez, E. Tapanari, M. Diekhans, F. Kokocinski, B. L. Aken, D. Barrell, A. Zadissa, S. Searle, I. Barnes, A. Bignell, V. Boychenko, T. Hunt, M. Kay, G. Mukherjee, J. Rajan, G. Despacio-Reyes, G. Saunders, C. Steward, R. Harte, M. Lin, C. Howald, A. Tanzer, T. Derrien, J. Chrast, N. Walters, S. Balasubramanian, B. Pei, M. Tress, J. M. Rodriguez, I. Ezkurdia, J. van Baren, M. Brent, D. Haussler, M. Kellis, A. Valencia, A. Reymond, M. Gerstein, R. Guigo, and T. J. Hubbard. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res., 22(9):1760–1774, Sep 2012. |
| [ZAA+18] | Daniel R Zerbino, Premanand Achuthan, Wasiu Akanni, M Ridwan Amode, Daniel Barrell, Jyothish Bhai, Konstantinos Billis, Carla Cummins, Astrid Gall, Carlos García Girón, Laurent Gil, Leo Gordon, Leanne Haggerty, Erin Haskell, Thibaut Hourlier, Osagie G Izuogu, Sophie H Janacek, Thomas Juettemann, Jimmy Kiang To, Matthew R Laird, Ilias Lavidas, Zhicheng Liu, Jane E Loveland, Thomas Maurel, William McLaren, Benjamin Moore, Jonathan Mudge, Daniel N Murphy, Victoria Newman, Michael Nuhn, Denye Ogeh, Chuang Kee Ong, Anne Parker, Mateus Patricio, Harpreet Singh Riat, Helen Schuilenburg, Dan Sheppard, Helen Sparrow, Kieron Taylor, Anja Thormann, Alessandro Vullo, Brandon Walts, Amonida Zadissa, Adam Frankish, Sarah E Hunt, Myrto Kostadima, Nicholas Langridge, Fergal J Martin, Matthieu Muffato, Emily Perry, Magali Ruffier, Dan M Staines, Stephen J Trevanion, Bronwen L Aken, Fiona Cunningham, Andrew Yates, and Paul Flicek. Ensembl 2018. Nucleic Acids Research, 46(D1):D754–D761, 2018. URL: http://dx.doi.org/10.1093/nar/gkx1098, arXiv:/oup/backfile/content_public/journal/nar/46/d1/10.1093_nar_gkx1098/2/gkx1098.pdf, doi:10.1093/nar/gkx1098. |